- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot  ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 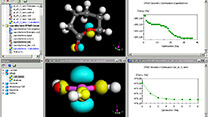 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
触媒分野特集
触媒研究者の皆様へ シミュレーションを活用しませんか?
- 触媒吸着反応などの実験の現象結果に対して、理論的な裏づけが得られます。(研究結果の検証)
- 触媒の実験で得られた現象を、計算機を使って解明し、新しいアプローチを創出する。(研究課題の深堀)
- 実験前に計算を行うことで、候補構造の絞込みが可能です。(実験の効率化)
- インパクトファクターの高い雑誌への掲載にご活用いただけます。(研究評価のアップ)
水分解反応における ZNO/GAP1-x Nxの光触媒活性
長岡技術科学大学の井上泰宣特任教授および東京大学の堂免一成教授らの研究グループとの共同研究により、ZnO/GaN母材の光触媒活性が解明されました。ZnO/GaN固溶体光触媒は、リン化することにより水分解の活性が著しく増強されます。リン化した ZnO/GaNの回折角度は、通常のGaPよりも2θ=0.20~0.44°高角側でGaPの単一のピークが発生しました。これはGaP1-x Nx合金系の形成を示しています。続いて、第一原理計算を使用して、Ga32P32-yNyの分子モデルで回折ピークのシミュレーションを行いました。実験におけるピーク・シフトとの比較により、GaP1-x Nx の X の範囲が0.034~0.074であるとき最も高い活性が誘導されることがわかりました[1]。
序論
可視光で活性化される新たな水分解光触媒が大きな進展を遂げています。d10型の電子配置を持つ、典型元素で構成される光触媒の中でもGaNは有望な候補の1つです。RuO2と組み合わせると水分解に活性になります[1]。GaPはd10電子状態を持つリン化物です。この材料のバンド・ギャップは300Kでおよそ2.25 eVであり、光吸収しきい値はおよそ560nmです。しかし、これまで水分解光触媒としての応用はわずかしか報告されていません。少量のNをGaPに含めると、GaPの間接遷移(T15-X1)[2]から直接遷移への基礎的な光学遷移の変化と、Nの密度向上によるバンド・ギャップの低下[3]が引き起こされることが多くの報告で示されています。Nが誘導するバンド・ギャップの低下は、窒素原子とGaP母材間の相互作用(GaP1-x Nx合金系を形成)の結果です[4]。これらの結果は GaP1-x Nx化合物、あるいは少なくともGaNまたはGaPでのGaP1-x Nx層形成が効率的に可視光吸収による光励起キャリアを生成することを示しており、GaP1-x Nx系を水分解の光触媒として応用することが期待されています。
この研究は実験とコンピュータ・シミュレーションを組み合わせた事例です[1]。以下のページでは、シミュレーションの結果に注目し、必要に応じて実験を参照しています。実験の詳細については文献[1]を参照してください。
モデルと手法
実験用のXRDパターンと比較する結晶構造のシミュレーションでは、64原子(Ga32N32)から成る2x4x1のスーパーセルを使用しました。このモデルは、ドーパントであるリンの対称な代替物を提供するために選択されました。周期的境界条件を考慮し、ドーパントの相互作用を最小化するために中央のストランドは除去されました。
計算はすべてBIOVIA Materials Studio(ダッソー・システムズ・バイオビア)のCASTEP(Cambridge Serial Total Energy Package)と、関連する対称性解析プログラムを使用して行われました。これは参考文献[1]で詳しく説明されています。XRDシミュレーションは、BIOVIA Materials Studio(ダッソー・システムズ・バイオビア)のReflexモジュールを使用して行われました。
目的
適切な材料を合成することが一番の課題です。合成した材料はキャラクタリゼーションを行う必要があります。光触媒活性はN/P比に依存することが実験的に観察されています。Pの位置は水の光触媒活性にとって非常に重要ですが、GaN内のPの位置を実験から知ることは非常に困難です。そのため、材料の特性と構造を合理的に説明するために分子モデリングを使用しました。
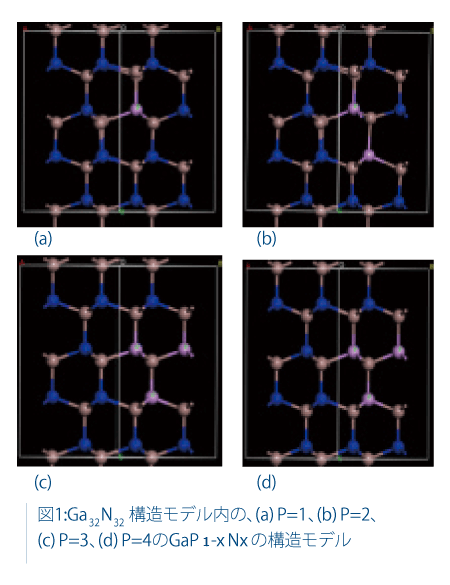
結果
PをドープしたGaNの望ましい構造を得るために、複数の合成法を試行しました。873~1123Kの窒化GaPのX線回折パターンは、GaPの強いピーク複数から成り、有意なピーク・シフトは見られませんでした。また、GaNに起因する非常に小さなピークが見られました。窒化GaPに担持させたRuO2 については、極めて少量のH2の生成がありましたが、それほど著しいH2およびO2の生成はありませんでした。
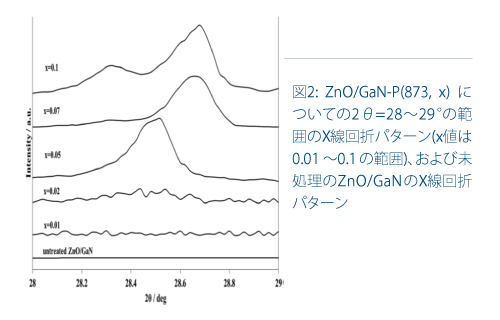
図2は、あるZnO/GaN-P(873,x)についてxを変化させたときの2θ=28-29°の領域のX線回折パターンを示しています。x=0.05では単一のピークが2θ=28.50°に見られ、x=0.07で2θ=28.68°にシフトしました。x=0.1については2θ =28.32°と2θ = 28.68°の2つのピークが観察されました。GaPのピークの高角度へのシフトは、N元素が四面体GaP4の基本単位に影響を及ぼし、GaP1-x Nxを生成したことを示しています。これはN3-のサイズがP3-よりも小さいためです。すなわち、GaPの回折ピークのシフトは、Pの導入が誘導する活性点の立体構造の変化を表しています。
回折角度が2θ=28.32°(GaP結晶)よりも大きくなるにつれて、823Kの光触媒活性が向上し、およそ2θ=28.6°で最大となり、回折角度がさらに大きくなると低下します。類似の傾向が873Kのリン化でも見られます。
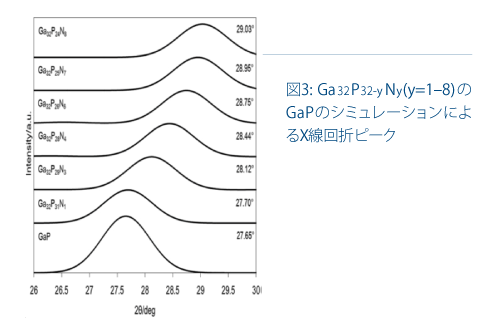
これらの相関は、高い光触媒活性が、PとGaNの気固反応を通じて形成されたGaP1-x Nxの構造に依存することを示しています。活性点の立体構造を究明するために、GanPn-yNyの構造最適化モデルを考慮して回折ピークのシフトのシミュレーションを行いました。図3は、BIOVIAのReflexモジュールに実装された粉末回析法で計算されたGaPの回折ピークを示しています(n=32およびy=1、3、4、6、7、8)。2θはステップ幅0.05で10~90°の範囲をプロットしました。ピーク位置の誤差は、ゼロ点が不正確であるか、試料の配置または透過性に関わる誤差に起因します。このシミュレーションでは、Bragg-Brentano補正を使用しました。Ga32P32のピークは2θ=27.65°で、N原子含有量が増加すると高角度にシフトし、y=4で2θ=28.44°、y=6で2θ=28.75°、y=8で2θ=29.03°に達しました。P原子とN原子の合計に占めるP原子の割合(P/(P+N)x100%)に対する回折角度のシフトは良好な直線関係を示しました。
GaPから高回折角度側に2θ=0.20~0.44°シフトすると最高の光触媒活性が得られた実験値と比べ、P原子とN原子の合計に占めるP原子の割合(P/(P+N)x100%)は、良好な直線関係を示しました。
この直線関係から、最も高い活性の最適条件はGaPにNを3.4~7.4%の割合で含めること、という結果が得られました。ZnO/GaN-Pの価電子帯はGa 4p+P 3p+N 2pの混成軌道からなり、GaPおよびGaNの中間バンド構造を形成します。このように、N原子をGaPに含めると、価電子帯端が、酸化が進むのに十分な水準まで低下します。これは水の酸化に対して正の影響がありますが、伝導帯端はより低いエネルギー準位にシフトします。
結論
活性なZnO/GaN光触媒のリン化は水分解の光触媒活性を著しく増強します。ZnO/GaNのリン化により、GaP1-x Nxにおける最適な窒素の環境を達成することが可能になり、高い光触媒性能の発揮に繋がります。この予測手法は他の金属窒化物系への応用が有望であり、効率的な水分解光触媒の実現に有益な手法となるでしょう。
BIOVIAは、アメリカ合衆国、またはその他の国における、ダッソー・システムズまたはその子会社の登録商標または商標です。
参照文献
1. C.Oshima,H.Nishiyama, A. Chatterjee,K.Uchida,K.Sato,Y.Inoue,T.Hisatomi and K.Domen, J. Mater.Chem.A, 2015, 3, 18083-18089
2. J. R. Chelikowsky and M. L. Cohen, Phys.Rev. B, 1976, 14,556–582
3. S. Miyoshi, H. Yaguchi, K. Onabe, R. Ito and Y. Shiraki, Appl.Phys.Lett., 1993, 63,3506–3508
4. J. N. Baillargeon, K. Y. Cheng, G. E. Hofler, P. J. Pearah and K. C. Hsieh, Appl.Phys.Lett., 1992, 60, 2540-2542.
ダッソー・システムズ・バイオビア社 チャタジー氏が参画した論文リスト
Synthesis(合成)
- Computer simulation studies on the role of templating organic molecules in the synthesis of ZSM-5. JCS Faraday Trans. 91 4313-4319.
- Simulation studies on the role of templating organic molecules in the synthesis of microporous materials :2. Modelling electronic interaction between the templating molecule and ZSM-5. J. Mol. Catal. 106 75-81.
- A computational expert system approach to design synthesis routes for zeolite catalysts. Stud. Surf. Sci. Catal. 105A 133-140.
- The effect of nature and number of alkyl groups in tetraalkyl ammonium cations acting as templates during the synthesis of ZSM-5. J. Mol. Catal. 120 155-163.
- A Reactivity Index Study to Choose the Best Template for a Particular Zeolite Synthesis, J. Phys. Chem. A 105, 6187-6196.
- A Reactivity Index Study to Choose the Best Template for Zeolite Synthesis, Stud. Surf. Sci. Catal. 135, 265-272.
- Synthesis and arsenic adsorption capability of smectite-titanium oxide nanocomposite of tunable pore size, Clay Science 12, 85-89.
- Comparative behavior of Pd and Ag deposition phenomenon on clean α-Al2O3 (001) surface ·A First Principle Study; Molecular Simulation 32 (2) 153-162.
- Au-Nano-particle deposition on oxide surfaces to tune a novel material for catalytic application – a first principle study., International Journal of Advanced Microscopy and Theoretical Calculations Letters, 1, 200-201.
- Preparation of Silica Sphere with Porous Structure in Supercritical Carbon Dioxide Journal of Colloid and Interface Science 348 (2010) 57–64
- Photocatalytic activity of ZnO/GaP1−xNx for water splitting (2015) J. Mater. Chem. A, 2015, Advance Article 2015, 3, 18083
ADSORPTION(吸着)
- Density functional calculation on the adsorption of nitrogen oxides and water on ion-exchanged ZSM-5, Appl. Surf. Sci. 130-132 561-565.
- A comparative study of DFT and XPS with reference to the adsorption of cesium ions in smectites, Computational Mater. Science 14 254-260.
- A novel way to design suitable inorganic material from smectite family for sorption of 2,3,7,8-tetrachlorinated Dibenzo-p-dioxin, J. Phys. Chem. A 104 2098-2104.
- Adsorption and Structural Energetics of Chemisorbed F atom on Si (100) – a Density Functional Theory Study, Japanese Journal of Applied Physics 39 4279-4284.
- Best dioctahedral smectite for nitrogen heterocyclics adsorption – a reactivity index study, J. Phys. Chem. A 105, 10694-10701.
- Adsorption structures and energetic of fluoro- and chlorofluorocarbons over faujasite – a first principle study, Stud. Surf. Sci. Catal. 145, 371-374.
- Intermolecular Reactivity Study to Scale Adsorption Property of Para and Meta Substituted Nitrobenzene over 2:1 Dioctahedral Smectite, J. Chem. Phys. 118, 10212-10220.
- Cholorofluorocarbons adsorption structures and energetic over faujasite type zeolites – a first principle study, THEOCHEM 630, 233-242.
REACTION(反応)
- Activity of C-H and C-C bond in the cracking reaction over isomorphously substituted ZSM-5 - a density functional study, Material Research Society Proceedings of 12th IZC, Edited by M.M.J. Treacy, B. Marcus, J.B. Higgins and M.E. Bisher, 489-496.
- Computer simulation study on acylation reaction of aromatic hydrocarbons over acidic zeolites, J. Catal. 185 23-32.
- The role of solvent on selective hydrogenation of conjugated and isolated C=C of citral (3,7-dimethyl 2,6-octadienal) - a self consistent reaction field study, Chemical Physics Letters, 395, 143-149.
- Switching of PET Fluorescence signals induced by ligand exchange reactions of N-(9-Anthrylmethyl)amine on Cu(II) complexes and its application to postcolumn detection of glyphosphate, Analytical Sciences 21, 417-420.
- Rapid Hydrogenation of Aromatic Nitro Compounds in Supercritical Carbon Dioxide: Mechanistic Implications via Experimental and Theoretical Investigations". Advanced Synthesis and Catalysis. 2012, 354, 2009 – 2018
- Caltalysis Science & Technol Rhodium-mediated hydrogenolysis/hydrolysis of the aryl ether bond in supercritical carbon dioxide/water: an experimental and theoretical approach ; Catal. Sci. Technol., 2015, 5, 1532.
- Dehydrogenation of 5-hydroxymethylfurfural to diformylfuran in compressed carbon dioxide: an oxidant free approach . Green Chem., 2017, 19, 1315-1326
国内研究者による論文
※ 論文タイトルをクリックすると、論文が掲載されているサイトに遷移します。(敬称略・新着順)
触媒分野向け製品のご紹介
合成
- 有機分子のコンフォメーション解析に!【MS Conformers】
強力な解析機能との連携により、分子のコンフォメーション空間に関する貴重な情報を提供し、低エネルギーコンフォメーションの同定を支援します。Conformersはコンフォメーション空間の網羅的な探索データを集め、分析する手法を提供します。単純なものから複雑な系まで、種々の系に応用が可能です。 - 分子力学、動力学計算なら!【MS Forcite Plus(MS COMPASS)】
多様な系における分子動力学研究を支援する、経験的分子軌道法にもとづく分子動力学計算ツールです。分子力場・凝集エネルギー密度の計算が可能。 - 粉末X線スペクトル予測に!【MS Reflex】
- モデリングツール【MS Visualizer】
分子や結晶、ポリマーなどのグラフィカルなモデルを作成し、モデルを操作し、表示観察し、解析することができます。また、グラフ、表、テキストを扱う機能も備えています。
反応合成
- 第一原理計算なら!【MS DMOL3 SolidState】
密度汎関数理論(DFT)に基づく量子力学プログラムです。気相、溶液、および固体におけるプロセスの予測が可能。均一触媒作用、不均一触媒作用、半導体、分子反応性と燃焼テクノロジーなどのリサーチなど、化学、薬学、物質科学、化学工学、固体物理学での課題の研究に幅広く応用できます。 - 第一原理計算、EELSスペクトル予測なら!【MS CASTEP】
密度汎関数法(DFT)によるab initio量子力学計算プログラムです。セラミックス、半導体、金属 を含む広範囲な材料の固体、界面、表面の物性をシミュレートするものです。第一原理計算では、構成原子の原子番号以外には何ら実験による情報は必要なく、 系の電子的、光学的、構造的特性とその起源を探ることができます。 - モデリングツール【MS Visualizer】
分子や結晶、ポリマーなどのグラフィカルなモデルを作成し、モデルを操作し、表示観察し、解析することができます。また、グラフ、表、テキストを扱う機能も備えています。
吸着
- MS Adsorption Locator
ゼオライト、カーボン ナノチューブ、シリカゲル、活性炭素などの幅広いマテリアルにおいて、 最も安定している吸着サイトを探索。 - MS Sorption
結晶性物質中の分子の吸着を予測するためのソリューション。吸着等温線(あるいは供給曲線)やヘンリー定数などの基本的特性を予測する手段を提供します。 - MS ForcitePlus
多様な系における分子動力学研究を支援。経験的分子軌道法にもとづく分子動力学計算ツール。分子力場・凝集エネルギー密度の計算が可能。 - MS COMPASS
バルク状態の高精度な分子計算を目的として最適化された第3世代力場 - モデリングツール【MS Visualizer】
分子や結晶、ポリマーなどのグラフィカルなモデルを作成し、モデルを操作し、表示観察し、解析することができます。また、グラフ、表、テキストを扱う機能も備えています。
お問い合わせ
製品内容・納入実績について
Materials Studioは、国内300名以上の先生方にお使いいただいてます。製品内容や納入実績のご説明をご希望の方は、下記よりお問い合わせください。
製品デモについて
BIOVIAの技術者と共に、製品のデモンストレーションにお伺いいたします。ご希望の方は、下記よりお問い合わせください。
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)