- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot  ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 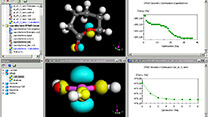 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
DFTB+
電子特性研究のための量子シミュレーションソフト
DFTB+は、物質中の電子特性を研究するための量子シミュレーションソフトウェアであり、従来のタイトバインディング法を改良し、密度汎関数理論をもとに定式化した手法が実装されています。DFTB+は密度汎関数理論に基づく第一原理計算に匹敵する精度と半経験的手法に迫るスピードを両立することができ、多数の原子を含む系を研究、分析する際に他に類を見ないほどの性能を発揮します。半導体の欠陥や有機/無機界面の相互作用など、これまでは時間やコンピュータの計算能力の面で大部分の研究者が対応できなかった問題も、DFTB+を使うことで研究が可能になります。こうした問題は、触媒、電子、化学などのさまざまな分野に共通して見られるものです。
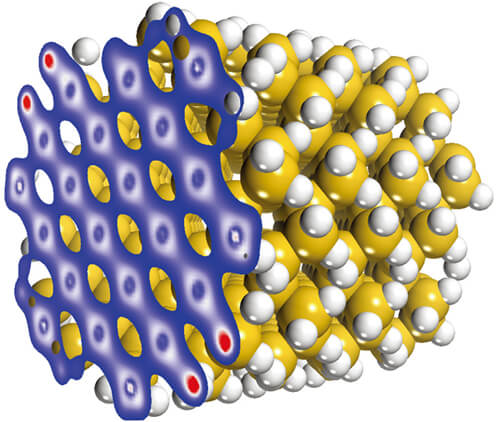
機能
DFTB+を使用することで、構造の最適化や動的な物性の研究において、第一原理計算に匹敵する精度の計算を今までにないほど短時間で実行できます。例えば、構造を最適化した後、分子動力学を使ってその構造の経時変化を追跡することが可能です。また、バンド構造、分子軌道やフェルミ面を計算し可視化すれば、物質の電子構造を深く理解できます。原子電荷の解析や電子密度分布の可視化により、電荷分布に関する知見を得ることが可能です。 DFTB+はSlater-Kosterファイルと呼ばれる元素間の相互作用をまとめたパラメータライブラリを使います。元素がパラメータ化されていない場合、DFTB+では系や目的に応じたパラメータ作成を可能にするタスクを追加で実行できます。それによりオリジナルのパラメータを作成し、新しい系への拡張を可能にします。
主な用途
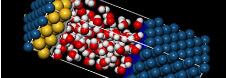
数千の原子を含む系をモデル化することで、半導体を扱う研究者は、1%以下という微量の欠陥が系に及ぼす影響を研究することができます。
分子層と表面の相互作用、および分子層間の相互作用、この両方を考慮した計算を行うためには、電子状態を正確に記述するだけでなく、大量の原子を扱うことができる手法が必要です。DFTB+はこの要件を満たしており、第一原理計算では計算コストがかかりすぎる場合や、古典力場計算では必要な精度や物性が得られない場合にも有効です。
ダイヤモンド表面上の窒化ホウ素など、無機結晶の成長を考察する際には、化学結合の生成と切断を電子状態の変化として取り扱い、しかも大規模な系においてこの計算を行う必要があります。DFTB+の電子状態計算は正確で、かつ大規模な系でも実行可能です。そのためより現実的な結晶成長のシミュレーションが可能です。
DFTB+ の仕組み
その上で、経験的近似を導入することにより、精度を維持しながら、計算速度の向上を達成しています。DFTB+で主に使用される近似は、DFTの厳密な多体ハミルトニアンを、パラメータ化されたハミルトニアン置き換えるというものです。DFTB+では、Slater型軌道と球面調和関数を用いて記述された疑原子軌道関数によって電子密度がモデル化されます。原子軌道基底は各元素について1原子をDFTで解いて得られたものであり、その原子軌道底を使ってハミルトニアン行列要素と重なり行列要素が計算されます。また、DFTで記述された全エネルギーをMullikenの電荷とスピンのゆらぎの二次まで展開するという近似を行います。こうして得られた行列要素は系の全エネルギーを完全には表現していないため、残りの部分は短距離反発項として含められます。この短距離反発項は各元素間で適切に設定された2体ポテンシャルとして記述されます。このポテンシャルはDFT計算結果へのフィッティングにより決定されます。
計算を簡素化する目的で、SCC計算は、ポテンシャルや電荷密度ではなくMulliken電荷について行われます。通常の電子項と短距離反発項に加え、最終的なKohn-Shamエネルギーには、電荷ゆらぎによるクーロン相互作用が含められます。距離が離れている場合は、二つの点電荷の間の長距離静電気力となり、電荷同士が同じ原子内にある場合は、その原子における自己相互作用の寄与が近似的に含められます。 DFTB+はSCC法の実装により、従来のタイトバインディング法では適切な解が得られないような問題に対しても十分に適用することができます。
機能および性能
- 計算タスク
-
- エネルギー 1点計算
- 構造最適化(格子定数の最適化、および任意の分子を剛体とみなして構造最適化が実行できます。)
- NVE、NVT、NPH、NPTアンサンブルを使った分子動力学計算
- 新しいSlater-Kosterパラメータ ライブラリを作成するための、パラメータ作成タスク(パラメータは、DFTに基づく、LDA/PWC、GGA/PW91、GGA/PBE汎関数を使用してフィッティングできます。)
- すべてのタスクは非周期系と周期系に適用可能です。
- ファンデルワールス分散力補正が利用できます。
- スピン分極を考慮した計算が可能です。
- SCFの収束性を改善するためのSmearingが利用できます。
- パラメータセット
-
- CH – 炭化水素用パラメータ
- CHNO – 炭素、水素、窒素、酸素を含む分子用パラメータ
- SiGeH – シリコン、ゲルマニウム、水素を含む半導体用パラメータ
- 物性
-
- 状態密度
- バンド構造
- 電子密度の可視化
- フェルミ面の可視化
- 軌道エネルギーの計算と可視化
- Mulliken電荷の解析
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)