- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot  ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 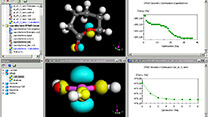 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
量子力学計算
DMol3の計算効率が大幅に向上しました。特に並列化効率が大幅に改善されました。また、ユニットセルの最適化の効率も向上し、分子性結晶などの構造最適化がより短時間で終了するようになりました。
電子輸送現象がDFTB+で取り扱えるようになりました。これにより、分子内の電子の透過率や、電流 - 電圧特性が計算できるようになりました。
TS Confirmation機能がCASTEPに搭載されました。これにより、反応経路のより深い理解が可能になります。
弾性定数をはじめとする、機械的物性がDMol3でも計算可能になりました。
古典力学計算
Forcite plusに溶解の自由エネルギーを計算する機能が搭載されました。
新しい力場パラメータ「COMPASS Ⅱ」が搭載されました。元となったCOMPASSの適用範囲をさらに広げたことに、イオン性分子やヘテロ環式化合物の計算がより高精度にできるようになりました。
Forcite plusにParticle-Particle Mesh Ewald法(P3M法)が搭載されました。これによりイオン性分子を含む大きな系の計算がより高速にできるようになりました。
Forcite plusにSouza -Martinsバロスタットが搭載されました。こにより、非等方的な応力を課した計算が可能になり、応力―ひずみ特性などの計算も可能になりました。
可視化機能とスクリプ
VisualizerにおいてGUIで設定した設定内容をMaterialsScriptとしてコピーする機能が搭載されました。こにより、簡単で間違いの少ないスクリプト作成が実現します。この機能はVisualizer内のほとんどのGUIに搭載されています。
Visualizerに電子輸送特性の計算のための充実したモデル作成機能が搭載されました。
Visualizerに平面と平面の間の角度を計測する機能、点と平面の距離を計測する機能が搭載されました。
システム環境
クライアントとサーバのOS環境として、Windows 8とWindows 8.1がサポート対象に追加されました。
TORQUE 2.5.13がLinuxにおけるキューイングシステムとしてサポート対象に追加されました。
Materials Studio 7.0新機能
- NEB 法を採用したTS ConfirmationタスクがCASTEPに追加されました。この機能により反応過程についてのより深い理解が得られます。
- Thermodynamic Integration法を使用した、溶媒やポリマー中の分子の溶媒和自由エネルギーの計算機能がForcite Plusに追加されました。
- 新しいCOMPASS II力場が追加されました。既存のパラメータに加えて、イオン液体のパラメータがのパラメータがイオン液体のパラメータが追加され、ヘテロ環化合物のパラメータが改善されました。
- Electron TransportタスクがDFTB+に追加され、透過率、I-V特性、電荷密度の計算が可能となりました。
- 電子輸送特性計算に使用する電極を分子や周期構造からの切り出しにより作成するための新しいツールが追加されました。Transport Deviceビルダーは電子輸送計算のための電極を含む完全なデバイスの構築を可能にします。複雑な伝導デバイスの作成が行えるようにLayerビルダーなどの既存のツールが電極のオブジェクトを利用できるように拡張されました。
Materials Studio 7.0改善点
- 明るい背景上のボールや円筒のオブジェクトに対してアンチエイリス処理が適用できるようになり、表示の品質が向上しました。
- 分子から平面への距離解析ができるようになりました。
- 2つの平面のなす角度の解析ができるようになりました。
- 構造、トラジェクトリ、スタディテーブルドキュメントを使ってMaterials Studioコレクションの過去のバージョンを利用しているPipeline Pilotサーバで計算を実行きるようになりました。
- エネルギー一点計算を行なうことなく、電荷の割り付けが可能となりました。
電子輸送計算のための電極のオブジェクトを取り扱えるようになりました。
- 構造構築のプロセスでのメモリ使用量が改善され、以前よりも大きな系の構築が可能となりました。
- 分子や表面に対する電場の効果を解析できるようになりました。
- 分散力補正の既存パラメータの修正や、新しい元素の追加、および交換相関汎数の適用範囲が拡張され、分散力補正の適用性が拡張されました。
- 双極子補正を行った計算のために2つの仕事関数が計算できるようになりました。
- TD -DFTを使って分子の励起エネルギーが計算できるようになりました。励起状態の構造最適化も可能となりました。
- 分子、表面、および結晶における電場の効果を解析できるようになりました。
- 原子のセットに対して部分状態密度の計算ができるようなりました。
- 振動モードの計算が可能となりました。
- MPIによる並列化が可能となり、大きな系でのパフォーマンスが向上しました。
- Souza-Martinsバロスタットが追加され、分子動力学計算において異方的な応力を適用することができるようになりました。
- 小さな系の計算が高速化されました。
- 分散力補正パラメータの追加、編集が可能となりました。各種交換相関相互作用に対しても適範囲を拡張できます。
- 有機分子からなる単位格子の構造最適化の効率が改善されました。最適化ステップ数と計算時間の両方でパフォーマン向上が期待できます。
- Density preconditionerを使用することによりSCF収束が改善されました。
- 弾性定数や機械特が計算可能になりました。
- Hirshfeld電荷解析により近似的に結晶振動モードの赤外強度を計算可能になりました。
- COSMO溶媒表面をVisualizer上で等値面または溶媒表面を表すメッシュ点で可視化することができます。
- バンドギャップ、価電子バンド端、伝導電子バンド端がテキストに出力されるようになりました。
- 遷移状態計算の際に予め生成物と反応物を自動的に構造最適化する機能が使用可能になりました。
- 双極子補正を用いた計算で2つの仕事関数が計算されます。
- Souza-Martins barostatが使用可能になりました。これよにより分子動力学計算中に異方的な応力を印加可能になります。
- 応力を変化させながら複数回の分子動力学計算を行うスクリプトStressStrain.plが同梱されます。このスクリプトはScript Libraryから直接実行可能です。
- 静電相互作用の計算にParticle -Particle -Particle Mesh Ewald non- bond法を導入することで、多数の原子を含む電荷に偏りのある系の計算効率が改善されました。
- エネルギー一点計算を実行しなくても電荷割り付けできるようになりました。
- 一定の外力を加えて分子の拡散を評価できるようになりました。
- Atom basedまたはGroup basedを用いたファンデルワース相互作用の計算で、long range correction機能が追加され、デフォルト設定でオンになります。
- ファンデルワース相互作用のデフォルトの計算法がEwaldからAtom basedに変更されました。
- アモルファス材料向けの熱伝導計算機能が追加されました。
- Souza-Martins barostatが追加され、分子動力学計算中に異方的な応力を印加可能になりました。
- 静電相互作用の計算にParticle-Particle-Particle Mesh Ewald non bond法を導入することで、多数の原子を含む、電荷に偏りのある系での計算効率が改善されました。
- 一定の外力を加えて分子の拡散を評価できるようになりました。
- Generate HabitタスクとGenerate Face ListタスクがMaterialsScript APIに実装されました。
- 構造最適化のパフォーマンス向上ためDZP基底セットが追加されました。
- ファンデルワールス相互作用の改善ため第一原理分散力補正が追加されました。
- 光学吸収スペクトル計算により光学材料の特性を評価可能になりました。
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)