- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot  ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 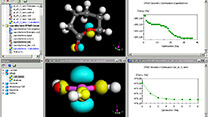 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
電子状態の計算精度の向上
- 金属の正確な電子状態計算のために、スピン軌道相互作用の補正やmeta-GGA汎関数が実装されました。
マルチスケールシミュレーションのワークフロー強化
- メソスケールシミュレーションのための力場の開発にはさまざまな技術が必要となります。Materials Studio 2016には、メソスケールでの相互作用の記述に必要となる、より柔軟な関数形を取り扱う機能とメソスケールシミュレーション用の力場の生成を自動化するためのツールが搭載されています。
半経験的量子力学計算手法の適用範囲の強化
- DFTB+はDFTの計算精度で比較的大規模な系を計算するための強力なツールですが、シミュレーションにはその系に含まれる元素のパラメータが必要となります。Materials Studio 2016には、有機物、ハロゲン化物、および生体分子向けの新しいより高精度なパラメータセットが含まれています。
古典力学計算のハイライト
- 結合、結合角、van der Waals相互作用に対してTabulatedポテンシャルが利用可能となり、 ポテンシャルの定義の自由度が非常に高くなりました。
- Iterative Boltzmann Inversion法に基づくメソスケールシミュレーション用の力場生成のためのスクリプトでTabulatedポテンシャルが利用できるようになりました。
量子力学計算のハイライト
- CASTEPでスピン軌道相互作用が計算可能となり、状態密度などの電子状態を高精度に計算できるようになりました。
- CASTEP NMR計算で スピン結合定数を計算できるようになり、材料を同定しやすくなりました。
- DMol3のB3LYP汎関数を使用した計算が改善され、大きな系にも適用しやすくなりました。
- DMol3のmeta-GGA汎関数が周期系にも適用可能となり、遷移金属元素などの重元素の計算精度が向上しました。
- DFTB+に3次の補正を考慮した新しいSlater-Kosterライブラリを追加。より幅広い元素に対応することで、結合エネルギーなどの計算精度が向上しました。
お気軽にお問い合わせください
各モジュールの機能強化
- Materials Visualizer
-
- Pipeline Pilotサーバーとの接続が高速化し、プロトコルの設定が自動的に保存される機能が追加されました。
- Properties Explorerにおいて、単純なプロパティ値をすばやく直接編集できるようになりました。
- CASTEP
-
- フォノンの状態密度計算から異方性温度因子が計算できるようになりました。
- On the flyで擬ポテンシャルを生成するときに、相対論的な効果に関する新しいオプションが追加されました。今までのKoelling-Harmonによる方法に加えて、ZORAと非相対論的な取り扱いが追加されました。これらの新しいオプションはスピン結合定数の計算で必要となります。
- 新しい構造最適化アルゴリズムとしてTPSD が実装され、格子の拘束をかけた構造最適化がより速く収束するようになりました。
- TD-DFTによる光学スペクトル計算が固体の計算にも拡張されました。
- 弾性定数計算で音速の平均値とデバイ温度が計算できるようになりました。
- 新しいノルム保存型のOn the fly擬ポテンシャルが利用可能となり、ノルム保存型の擬ポテンシャルにのみ対応している計算機能 (例えば線形応答理論、非局所型汎関数などを使用する計算)の計算精度が向上しました。
- CASTEP NMR計算でスピン結合定数を計算できるようになり、材料をより同定しやすくなりました。
- ServerAtomNameプロパティにより原子がラベル付けされ、CASTEPのテキストファイルと3Dモデルの原子を対応付けることができるようになりました。
- Ybのノルム保存型の擬ポテンシャルが追加されました。
- Jobの完了時にエネルギーや収束などのチャートが自動的に更新されるようになりました。
- 内殻励起スペクトルのチャートの解像度を設定できるようになりました。
- DFTB+
-
- 3次のDFTB法を使用する3obライブラリがDFTB+に追加されました。このライブラリは、C, H, N, O, P, S, Mg, Zn, Na, F, K, Ca, Cl, Br, Iの各元素に対応しており、デフォルトのライブラリとなりました。
- 金と有機物からなる系を対象とするパラメータセットが追加され、電気伝導特性などの計算に応用できるようになりました。
- GaN中の希土類元素の欠陥やハロゲン分子の計算に適したパラメータセットが追加されました。
- 電子伝導特性の計算の並列化効率が約30 %向上しました。
- DMol3
-
- 新しいmeta-GGA汎関数、MSx、TPSS、revTPSSが追加されました。また、meta-GGAが周期境界条件を課したモデルの計算にも対応しました。
- B3LYPの計算性能が著しく向上し、メモリの使用量も減少しました。
- 弾性定数計算で音速の平均値とデバイ温度が計算できるようになりました。
- 電子伝導計算タスクで電子密度と静電ポテンシャルの計算が可能となりました。
- 複雑な構造に対する構造最適化の収束性能が改善しました。
- エネルギー一点計算で各原子に働く力を返すオプションが追加されました。
- Forcite
-
- van der Waals相互作用のスケールファクターが導入され、OPLSなどの力場に適用することができるようになりました。
- 結合、結合角、van der Waals相互作用に対してTabulatedポテンシャルが利用可能となり、ポテンシャルの定義の自由度が非常に高くなりました。
- GULP
-
- Ni-TiのパラメータがMEAM-2nnに追加されました。
- Au-SiのパラメータがMEAM-1nnに追加されました。
- Mesocite
-
- 結合、結合角、van der Waals相互作用に対してTabulatedポテンシャルが利用可能となり、ポテンシャルの定義の自由度が非常に高くなりました。
- Iterative Boltzmann Inversion法に基づくメソスケールシミュレーション用の力場生成のための新しいスクリプトがExamples/Scriptingフォルダに追加されました。
- ONETEP
-
- 分子動力学計算が可能になりました。
- 金属の計算の収束性が改善しました。
- Reflex
-
- 規格化を行っていない回折パターンを生成できるようになりました。
- VAMP
-
- MaterialsScriptを使ったVAMPの計算において生成熱が得られるようになりました。
活用分野に最適なMaterials Studio製品をご紹介



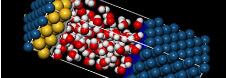
お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)