- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot 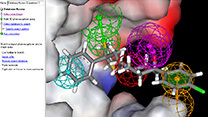 ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 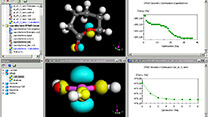 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
有機分子をはじめ、無機、高分子ポリマー、結晶構造、界面構造等を簡単に作成・編集可能。モデリング~化学計算~検討の研究サイクルを加速できます。
Materials Studioは、医薬品・触媒・ポリマー・混合物・金属・合金・電池・燃料電池など、さまざまな材料の研究開発にご利用いただける、分子モデリング・シミュレーションツール群です。専門分野に特化した20種類以上のソフトウェアの中から、お客様の研究領域にあわせてご導入いただけ、統合された操作環境でご利用いただけます。複雑な構造計算をする前におおまかな構造検索が行え、研究を効率化できます。スペクトル予測(NMR,IR,Raman、X線)も可能です。



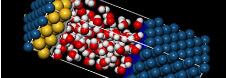
最新論文
敬称略
広島大学 松田海斗、教授 犬丸啓
Materials Studio販売実績
大学
- 北海道大学
- 東北大学
- 東京大学
- 東京工業大学
- 北陸先端科学技術大学院大学
- 名古屋大学
- 奈良先端科学技術大学院大学
- 京都大学
- 大阪大学
- 九州大学
- 他、日本国内大学計算機センターおよび研究室へ販売実績多数
官公庁
- 産業技術総合研究所
- 理化学研究所
- 物質・材料研究機構
- 他、日本国内官庁系研究機関への販売実績多数
企業
- 日本国内の企業研究機関への販売実績多数
Materials Studio販売の歴史
- 1985年 自社開発の分子シミュレーションソフトウェア「MOLーGRAPH」を販売開始
- 1996年 Biosym社の販売店として、InsightII、Cerisu2(Materials Studio 前身ソフト)の販売開始
- 2000年 Cerius2の後継システム Materials Studio1.0発表(Materials Studio販売開始)
- 2001年 Accelrys社が設立され、Accelrys社の販売代理店として Materials Studioの販売を継続
- 2018年 ダッソー・システムズ社の販売代理店として、Materials Studio販売を継続、現在に至る
お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)