- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot  ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 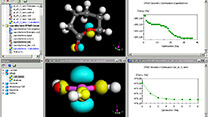 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
金属材料向けソフトウェア
MS Visualizer 有機~無機結晶までを構築できる分子モデリングツール
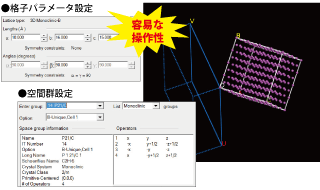
Crystal, Surface, Layer 構築 ツール
- 格子パラメータ、原子座標と対称性(空間群)に基づいて周期系構造を作ります。
- 広範囲の空間群情報を含む対称性の入力や編集機能。
- 周期系モデルを解析し対称性の詳細情報を表示します。
- 対称性に基づいて非周期構造から簡単に表面構造を作成。
- 真空スラブと共に結晶構造から簡単に表面構築を作成。
- 結晶、非結晶マテリアル、或いはそれらの組み合わせから層状構造を構築できます。
Polymer Building ツール
- ホモポリマー、ブロックポリマー、ラ ンダム共重合体やデンドリマーを構築できます。
- 繰り返し構造(repeat unit)を持った広範囲のライブラリ。
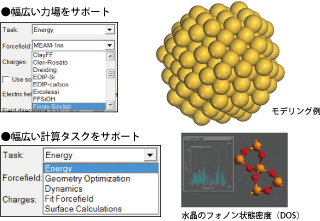
MS GULP 分子力学/分子動力学シミュレーション(MM/MD)
最新の古典力学計算ツールであり、エネルギー計算/構造最適化/動力学シミュレーションが可能。
金属、結晶など広い適用性を備えています。
活用分野
- 酸化物表面での不均一触媒系 ・ 燃料電池
- 核廃棄物処理プロセス ・ 水蒸気電気分解 ・ ガスセンサ
- 自動車排ガス触媒
- 石油化学
- ガラス
GULPで計算できる物性
- Properties of oxides
- Point defects, impurities & interstitials
- Surface properties
- Migration of ions
- Reactivity and Structures of zeolites and other microporous materials
- Ions intercalated in clays
- Properties of ceramics
- Disordered structures

お気軽にお問い合わせください
MS CASTEP 第一原理計算(ab initio)プログラム
平面波基底関数(Plane wave basis function)を用いた分子、固体、表面および界面の電子状態解析が可能

セラミック、半導体、金属などの幅広い材料に対して、固体、界面、および表面の特性をシミュレート。第一原理計算を利用することで、実験に基づいたパラメータを入力することなく、系の電子、光学、および構造の特性に関する本質と起源を調べることができます。
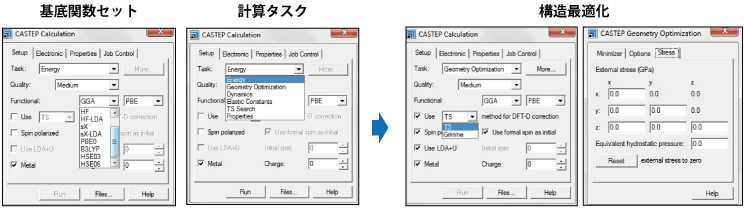
- LDA/LSDA/GGA(PW91, PBE)/ HSEなどによる全エネルギー、力とストレス
- 構造最適化(原子座標およびセル定数)
- 分子動力学(MD)、NVE/NVT/NPH/NPTアンサンブルを指定
- 遷移状態の探索(LST/QST/CG法)
- 弾性定数ならびに機械的性質(ヤング率、ポアソン比等)
- IRおよびUVスペクトル、光学的誘電定数
- 分極計算(誘電定数の格子からの寄与,Born有効電荷,分極率)
- Mulliken population解析(Mulliken電荷、結合次数)、信頼性を定量的に示すsplling parameterも出力
- 周期表全体に対するウルトラソフトおよびノルム保存(型)擬ポテンシャル
特性
- 光学特性:周波数依存の誘電関数分極率、反射率、屈折率、UVスペクトル
- フォノン分散、状態密度
- バンド構造
- 全体および局所フォノン状態密度
- 準調和近似に基づいた熱力学特性:自由エネルギ、エンタルピー、 エントロピー、熱容量、Debye温度)
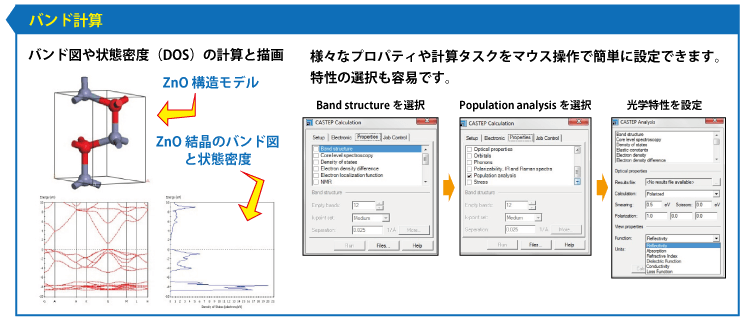
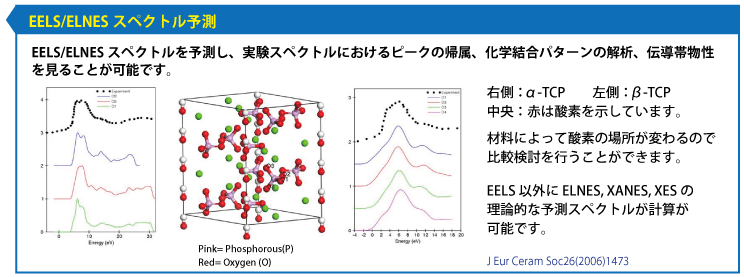
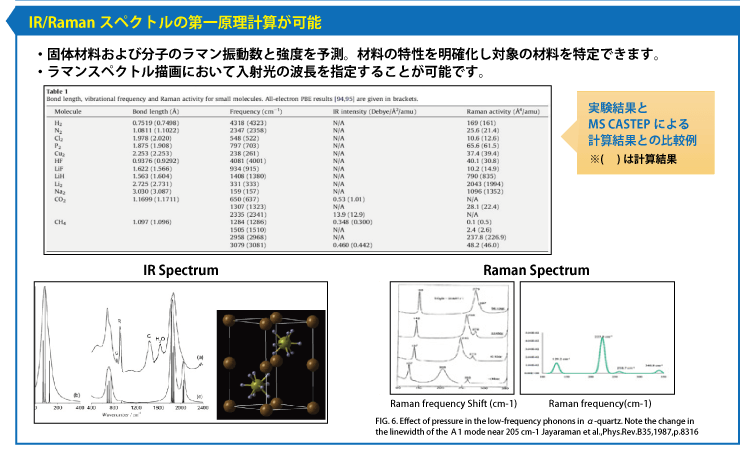
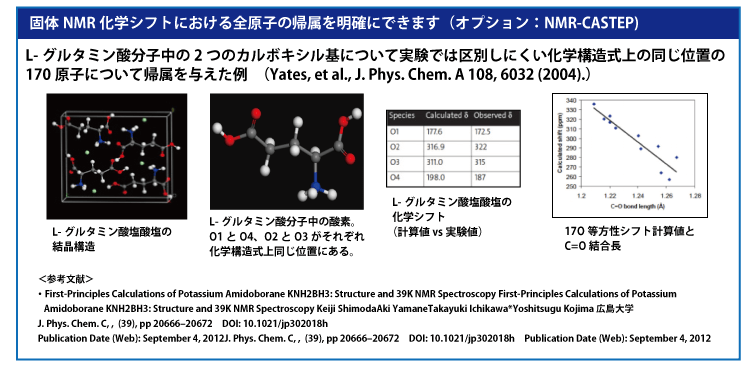
MS DMol3 密度汎関数理論(DFT)に基づいた量子力学プログラム
数値原子基底(Numerical atomic basis)により、大規模な系の計算が可能で構造、エネルギー、反応性などを予測
密度汎関数理論 (DFT) に基づいて化学プロセスをシミュレートし、物質特性を高速かつ正確に予測します。気相・溶液・固体におけるプロセスの予測が可能で、化学・薬学・物質科学・化学工学・固体物理学での課題の研究に幅広く応用できます。
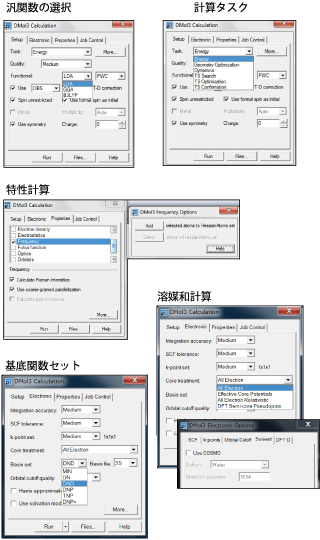
計算タスク
- スピン制限および非制限DFT計算
- エネルギー一点計算ならびに構造最適化
- LST/QST/CG(共役勾配法)法を組み合せた遷移状態探索
- 固有ベクトル追跡法(eigenvector following )による遷移状態構造の最適化
- 内部非局在座標系に基づく頑強な構造最適化アルゴリズム
- 完全あるいは部分(partial)Hessianを用いた振動数計算
- NEB(Nudged-Elastic Band)アルゴリズムに基づいた遷移状態経路の同定
特性
- Mulliken、Hirshfeld、およびESP電荷
- 静電ポテンシャル、双極子モーメント
- Fukui 関数
- Nuclear electric field gradients
- 結合次数解析
- 生成熱、自由エネルギー、エンタルピー、エントロピー、熱容量、零点振動
- 反応経路の可視化表示
- ラマン、IR、UVスペクトル計算
- COSMOによる溶媒効果
- 分子軌道、電荷、スピン/電子密度の可視化表示
- 多重k点(Multiple k-points、DMol3 Solidのみ)
- 様々なSCFオプション:DIIS,density mixing,smearing
- 状態密度(DOS)、部分状態密度(Partial DOS)
基底関数セット
- 数値原子軌道(AO)基底セット:Minimal、DN、DND、DNP、TNP、およびDNP+
- 全電子またはコアポテンシャル:相対論的有効コアポテンシャル、有効コア擬ポテンシャル、DFT semi-core 擬ポテンシャル
- スカラー相対論的な全ての電子計算
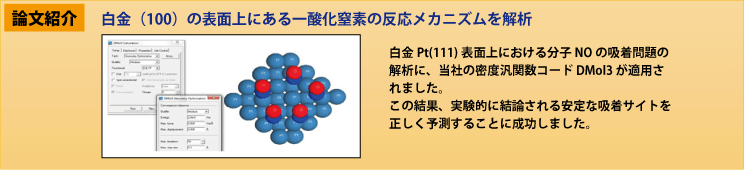
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)