- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot 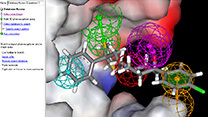 ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 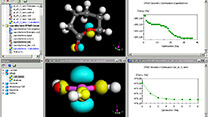 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
MORE SCIENCE – REACTION KINETICS
A new module, Kinetix, has been made available. Kinetix provides predictions for the spacial distribution of reactants and products on reactive surfaces, such as catalyst surfaces, as a function of time using a Kinetic Monte Carlo approach.
Tasks for examining the sensitivity of reaction mechanisms to individual reactions and for studying temperature programmed desorption have also been added to Materials Studio Cantera, to add yet more to the extensive suite of chemical engineering tools available in Materials Studio.
DENSITY FUNCTIONAL THEORY FOR 1000S OF ATOMS
The features of ONETEP available through Materials Studio ONETEP UI has been significantly enhanced by adding capabilities for electron and spin transport, time dependent DFT for electronic excitation energies, optimization of the total spin of metallic systems and extension of solvation models to geometry optimization, transition state search and dynamics tasks.
MORE MATERIAL PARAMETERS
A new DFTB+ library, LIB 2019, designed for simulations of electrolytes in Li-ion batteries, has been added. The library contains parameters for the elements Li, C, H, O, N, F, P and will allow Li-ion diffusion to be measured as a function of electrolyte formulation. New GULP libraries for MEAM and ReaxFF RDX are also made available which can be applied to battery and high energy materials respectively.
PERFORMANCE IMPROVEMENTS
Hybrid OpenMP/MPI parallelization has been made available for ONETEP and CASTEP calculations that will improve performance on compute clusters. Defaults have also been adjusted in ONETEP and Mesocite to improve performance without sacrificing accuracy. DMol3 geometry optimization in delocalized internal coordinates can also lead to substantial performance increases on highly parallel computers and/or for large systems, now that checkpoint information is no longer written to disk.
Low-memory BFGS method with preconditioning (LBFGS) has been implemented in CASTEP geometry optimizer. LBFGS calculations can reduce the number of optimization steps significantly, especially for systems with large number of degrees of freedom.
MORE PROPERTIES
Mulliken population analysis can now be applied in CASTEP for calculations with spin-orbit coupling. Additionally, prediction of chemical shifts are now possible for highly correlated systems where the use of DFT+U approach is required (for example, transition metal oxides or systems containing f-elements).
お気軽にお問い合わせください
MATERIALS STUDIO 2019のハイライト
Reaction Kinetics Extensions
【新機能】Sensitivity analysis has been added to Cantera CSTR task. This feature allows user to determine the most important steps in the reaction chain and potentially save time on simulations of complex multi-step reaction mechanisms by excluding less important reactions from needing to be considered. It is equally important to know which reactions merit more accurate calculations of thermodynamics and reaction barriers.
【新機能】A new task for simulation of Temperature Programmed Desorption has been introduced in the Cantera module.
【新スクリプト】A script has been developed to import third-party reaction mechanisms into Materials Studio to be used with Cantera.
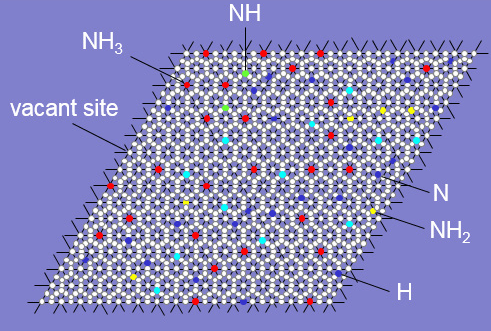
Figure 1. Image representing occupancy of Kinetix grid by adsorbate species during Kinetic Monte Carlo simulations
Quantum Mechanics Highlights
【機能強化】Finite displacement calculations of vibrational properties with CASTEP can be now performed using the “interpolation” option. This scheme uses non-diagonal supercells as developed by Lloyd-Williams and Monserrat and is more efficient than the previously available approach.
【新機能】CASTEP calculations of chemical shifts are now possible for highly correlated systems where the use of DFT+U approach is required (for example, transition metal oxides or systems containing f-elements).
【新機能】The many-body dispersion (MBD) correction scheme by Ambrosetti et al. (2014) has been implemented in CASTEP and offers a more accurate description of weakly bonded systems than other existing approaches. The MBD scheme supports full geometry optimization and property calculations.
【新機能】CASTEP output now reports the result of the check for “ghost states” that might be associated with the given pseudopotentials. If such states are found, results of optical properties calculations and band structure or DOS calculations of the conduction band will have spurious features. A number of settings for on the fly generation of pseudopotentials have been modified to eliminate ghost states.
【新スクリプト】A script for merging DFTB+ parameter files has been added to the DFTB+ Parameterization Tool Set available in the BIOVIA Materials Studio Community.
【新機能】The SCAN meta-GGA functional has been added to DMol3.
【新機能】A checkbox has been added to the geometry optimization and TS optimization dialogs in DMol3 to enable the use of Cartesian coordinates for optimization.
【操作性改善】DMol3 reporting of exchange-correlation functionals in the outmol file has been simplified and made more user friendly and reporting of the progress of a DMol3 Geometry Optimization or Transition State Optimization has been improved to explain why the process might continue even though the force and energy criteria are satisfied.
【新機能】Numerically stable Kohn-Sham eigenvalue solver has been added to enable solution of extremely rare systems where DMol3 detects a singular overlap matrix.
【操作性改善】QMERA calculations now allow you to set initial spins on atoms in the QM region for spin-polarized calculations.
【新機能】The ability to set global NGWFs radii for all elements has been added to the ONETEP interface.
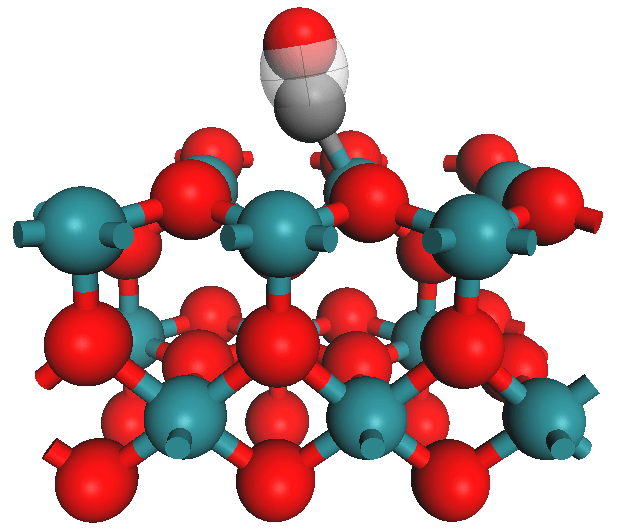
Figure 2. Image showing atomistic model of chemisorbed carbon monoxide on a RuO2(110) catalyst surface
お気軽にお問い合わせください
Classical Simulation Highlights
【新機能】The forcefield document, and Forcite and Mesocite, now support the tabulated form for torsion interactions.
【新機能】Forcite geometry optimization has been extended to allow for the optimization of a charged system which is subject to an electric field.
The latest version of GULP code, 5.1, has been incorporated in Materials Studio.
- New modified embedded atom model parameters for MEAM-2NN library to extend the range of accessible systems and MEAM-2NN-QEq, for modeling of Li-Mn-O materials relevant for simulations of batteries
- Split bond charge electronegativity equalization method (EEM) added
- Third party package Alamode, that allows calculation of thermal conductivity via the Boltzmann transport equations, can be connected GULP server.*
- Grüneisen parameters, phonon group velocities, mean squared displacements for phonon thermal parameters, eigenvalues of elastic constants for elastic stability check
- New potential forms added: Slater potential and buffered 14-7 Lennard-Jones potential
- Fitting of free energy and of absolute dipole moment
- Vibrational frequencies for molecules and (at Gamma point) for solids have been made available for ReaxFF and MEAM families of libraries
*注記: No Materials Studio interface to Alamode or third party software is provided.
【新パラメータ】The ReaxFF RDX library by Liu et al. (2011) has been added to GULP module to enable modeling of energetic materials.
Reflex
In Reflex Powder Solve and Powder Refinement, torsional degrees of freedom can now be restrained during refinement, by specifying a maximum variation.
Visualization and Usability Highlights
- File compression is now used by default for AVI movie export of trajectory animations from Materials Studio. This results in a typical file size reduction by a factor of ten. It is also observed that there are fewer playback problems with compressed AVI video than there had been for uncompressed AVI video.
- You can now browse a job folder of a running Pipeline Pilot protocol through a web browser, this is analogous to browsing gateway jobs.
- A new algorithm has been implemented for constructing Reaction Previews for 3D periodic systems.
- Structures with field density data can now be exported as Abaqus .inp models. The density fields are used to determine the material at each point, and elements in the Abaqus model are assigned to sets accordingly.
- The Pipeline Pilot Connector will now allow you to access some parameters from a protocol’s Implementation tab. This means that you can now use the Pipeline Pilot Connector to submit an entire protocol to a queuing system.
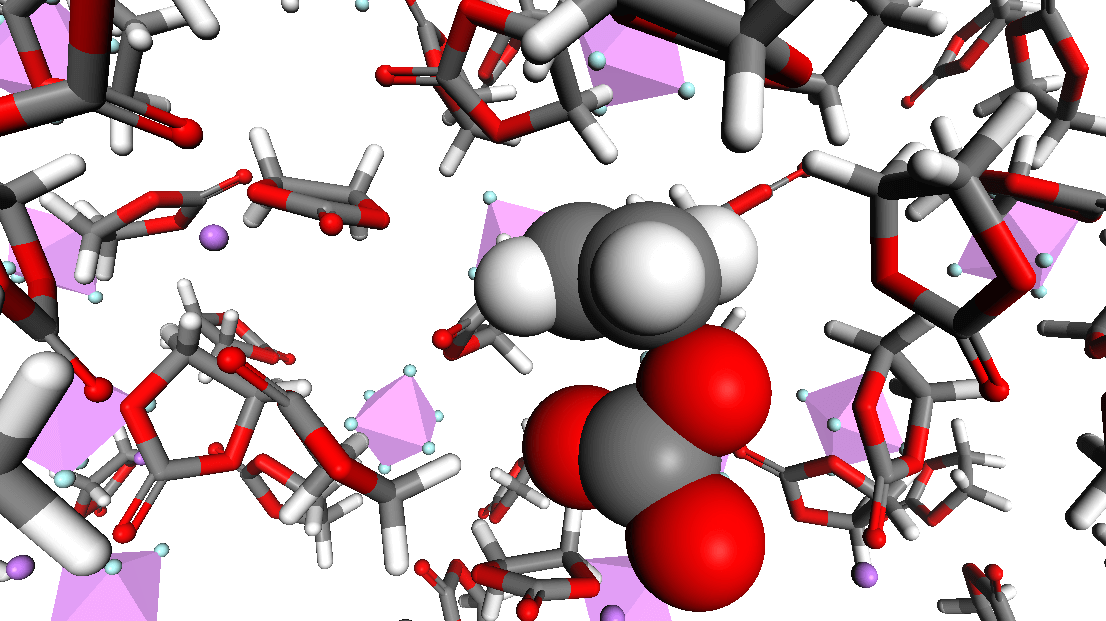
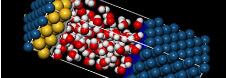
Figure 3. Image showing dissociation of ethylene carbonate during molecular dynamics simulation of a typical battery electrolyte formulation using LIB2019 DFTB+ library.
Enhancements to MaterialsScript
- Vector operations in MaterialsScript are now fully supported and documented.
- The ONETEP module is now scriptable with MaterialsScript.
お気軽にお問い合わせください
新チュートリアル
以下の新しいチュートリアルが追加されました。
- •Mechanism reduction using sensitivity analysis:
- Introduces the use of Cantera sensitivity analysis for studying reaction mechanisms. The problem-specific reduction of a reaction mechanism to contain only the reactions essential for the final outcome is presented.
- •Temperature programmed desorption:
- Introduces the use of Cantera for temperature programmed desorption.
- •Employing cluster expansion to calculate random alloy stability and properties using Pipeline Pilot:
- Demonstrates how to run the Random Alloy Stability and Properties (Cluster Expansion) protocol in the Materials Studio Collection for Pipeline Pilot from Materials Studio.
- •Calculating random alloy properties using Pipeline Pilot:
- Demonstrates how to run the Random Alloy Properties (Quasirandom Structures) protocol in the Materials Studio Collection for Pipeline Pilot from Materials Studio.
- •Cross-linking polymers using Pipeline Pilot:
- Demonstrates how to run the Create Polymer Network protocol in the Materials Studio Collection for Pipeline Pilot, from Materials Studio.
- •Calculating the stress-strain diagram using Pipeline Pilot:
- Demonstrates how to run the Calculate Yield Stress and Critical Distortional Strain protocol in the Materials Studio Collection for Pipeline Pilot from Materials Studio.
- •Calculating the glass transition temperature using Pipeline Pilot:
- Demonstrates how to run the Calculate Glass Transition Temperature protocol in the Materials Studio Collection for Pipeline Pilot, from Materials Studio.
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)