- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot  ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 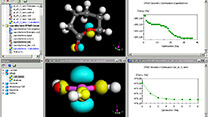 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio 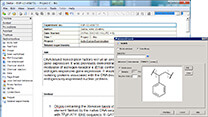 電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
BIOVIA Materials Studio 2022は、BIOVIAが化学・材料科学研究分野向けに提供する予測科学ツールの最新リリースです。Materials Studioは、材料の分子や結晶の構造と、さまざまなスケールで生じる特性との関係性を理解するために必要な機能を研究者に提供します。
BIOVIA Materials Studio 2022を活用する研究者は、原子からマイクロスケールまでを広範にカバーする、ワールドクラスのモジュール群やパラメータセットを利用することができます。本リリースでは、Materials Studio 2021で追加された2つのモジュールの機能を拡張・強化しました。1つはFlexTSの遷移状態探索、もう1つはPhaseFieldの硬質材料の微細構造シミュレーションです。その他にも、ForciteおよびMesociteでのCPUとGPUにおける古典シミュレーションの性能強化によるさらなる高速化や、Mesociteで新しい Martini 3の粗視化力場が利用できるようになりました。
BIOIVA Materials Studio 2022があれば、これまでにない精度と効率で、さらに多くの材料の特性解析が可能になります。
反応速度論に対応したBIOVIA MATERIALS STUDIO FlexTSワークフロー
BIOVIA Materials Studio 2021から、ケンブリッジ大学で開発されたプログラムをベースにしたFlexTSモジュールが導入され、最小エネルギー経路や遷移状態の計算、多段階反応の同定を含む、安定かつ効率的な反応経路計算手法が提供されるようになりました。BIOVIA Materials Studio FlexTS には段階的な計算手法が組み込まれており、まず最小エネルギー経路を同定し、次に遷移状態や個々の遷移状態に関連する中間状態を同定します。FlexTSの最小エネルギー経路計算手法は、BIOVIA Materials Studio DMol3モジュールとDFTB+モジュールのどちらからでも、Minimum Energy Pathタスクとして利用できます。
Materials Studio 2022では、以下の機能が強化されました。
- 性能の向上 - DMol3モジュールのプロセスを止めずにFlexTSによる最小エネルギー経路の計算が可能になり、DMol3の起動と完了のプロセスを最小限に抑えたことで、大幅に計算性能が向上
- 利便性の向上 - DMol3モジュールとDFTB+モジュールのMinimum Energy Pathタスクで以下の機能が追加
- - NEB 法により得られた中間状態のエネルギー分布の出力と反応経路の収束結果を容易に確認
- - 任意の中間状態のトラジェクトリファイルから計算を開始
FlexTSから取得した遷移状態に関する情報を活用して、DMol3モジュールのReaction Kinetics(反応速度論)タスクで速度係数を求めることができ、そのデータをBIOVIA Materials StudioのCanteraモジュールやKinetixモジュールで使用することができます。BIOVIA Materials Studio 2022を導入することで、化学に携わる企業や研究機関では、効率や安定性だけでなく、柔軟性にも優れた一連の計算手法を活用し、量子力学から化学反応速度論に至る、さまざまな研究をつなげることができます。
- 効率的かつ安定した動作で使いやすい遷移状態探索機能
- 反応経路の予測、遷移状態の最適化、活性化障壁の低い反応に対応
- Materials Studioの単一環境で複数のモジュール(DMol3、DFTB+、FlexTS、Kinetix、Cantera)を活用して、さまざまな反応を研究
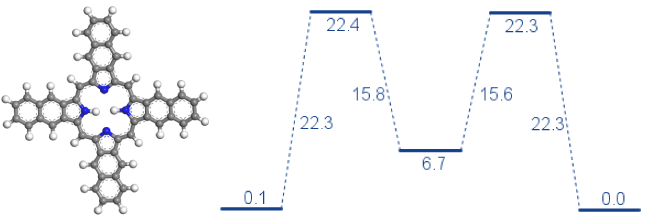
図1:FlexTSとDFTB+で計算された多段階反応と反応経路のエネルギーレベルの例 - Pipeline Pilot Materials Studio Collectionの専用プロトコルでFlexTSの結果を処理し、反応経路のグラフを作成
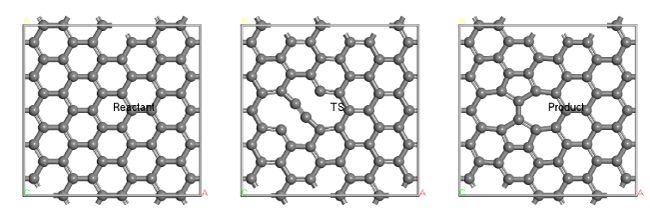
図2:グラフェンのストーン-ウォールズ欠陥生成における反応物・遷移状態・生成物
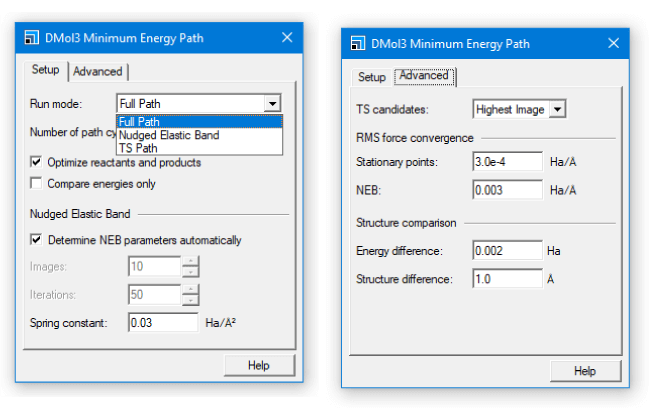
図3:DMol3で利用可能になった新しいタスクのダイアログ
- FlexTSを使用するMinimum Energy Pathタスクの設定が可能
合金設計に対応したBIOVIA MATERIALS STUDIO PHASEFIELDワークフロー
BIOVIA Materials Studio 2021から、OpenPhase CoreソルバーをベースにしたPhaseFieldモジュールが導入され、硬質材料の微細構造のシミュレーションが可能になりました。Pipeline Pilot Protocolsツールの使いやすいインターフェースを通じて、構成相や結晶粒子の設定、熱力学/動力学データの入力、温度/応力条件などを簡単に定義することができます。
こうしたシミュレーションの中でも重要なのが、金属積層造形の最適化が可能なアプリケーションです。パウダーベッド方式の積層造形において、微細構造の造形条件に対する依存性を把握するために、BIOVIA Materials Studio PhaseFieldを活用することができます。金属合金粉末の各層は、溶融・凝固することで結晶相が変化します。その結晶相には、冷却速度と局部温度の影響を受けやすいという性質があります。この依存性は、TTT図(Time-Temperature-Transformation Diagram)で示されます。Materials Studio 2022では、Pipeline Pilot Materials Studio Collectionの専用プロトコルでTTT図を作成できます。このTTT図の結果から必要なパラメータを読み取り、SIMULIA Abaqusを用いた積層造形プロセスの巨視的シミュレーションを実行することができます。
BIOVIA Materials Studio 2022を導入することで、材料科学や冶金学に携わる企業や研究機関では、原子スケールにおける合金混合の第一原理予測から、金属鋳造や積層造形の巨視的世界に至る、さまざまな研究をつなげることができます。
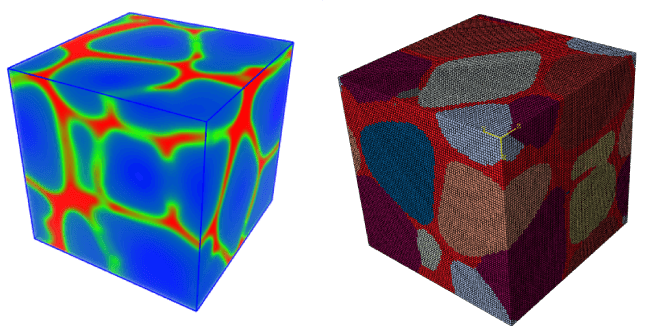
図4:BIOVIA Materials Studio PhaseFieldのシミュレーション機能で作成された2つの微細構造の粒界面を示す例 - 左側はBIOVIA Materials Studioによる結晶粒子分布の表示、右側はSIMULIA Abaqusソフトウェアによるボクセル表示
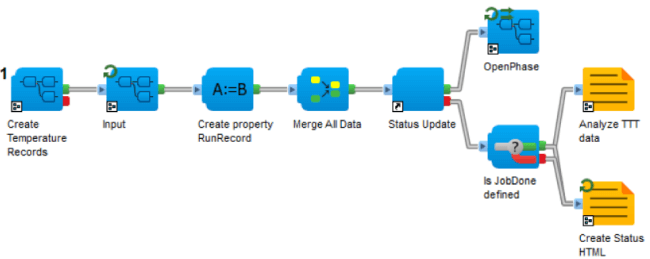
図5:Materials Studio PhaseFieldのシミュレーションは、Pipeline Pilotのプロトコルとして提供されています - フェーズフィールド法による一連の計算で構成されたTTT図のシミュレーションプロトコル
MATERIALS STUDIOモジュールの機能強化
古典シミュレーション性能強化とチュートリアル追加
Materials Studio 2021では、NVIDIAのGPU 環境に対応したことで、Forciteの古典分子動力学法に基づく高度な機能とMesociteの粗視化分子シミュレーションの性能が大幅に向上しました。Materials Studio 2022では、GPUとCPUの両方の性能をさらに強化し、Pipeline Pilot Materials Studio CollectionのForciteやMesociteを使用するプロトコルでも利用できるようになりました。
粗視化分子シミュレーションは、古典分子動力学法で使用する長さや時間スケールを拡大できることから、ますます普及が進んでいます。Materials Studio 2022では、新たに実装されたMartini 3力場を使用して精度の高い粗視化分子シミュレーションを実行して、多種多様な材料を分析できます。Martini 3に対応する新しいスクリプトツールにより、計算に使用する粗視化モデルやそのモデル専用のMartini 3力場の準備がさらに容易になります。詳細については、Materials Studioのヘルプ機能に新たに追加されたチュートリアルで学ぶことができます。
- パラメータの追加:MS Martini 3がMesociteで利用可能な力場に追加
- 利便性の向上:粗視化粒子ベース構造の準備ツールと、準備した粗視化モデル専用のMS Martini 3力場の作成ツール
- チュートリアルの追加:全原子のモデルから粗視化モデルを作成して分子のトポロジー情報を構築し、カスタマイズしたMS Martini 3力場を用いてMesociteのシミュレーションを実行する方法を説明します
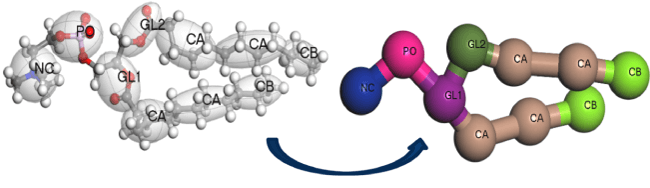
図6:Materials Studio 2022の新しいスクリプトツールを使用して、モデルの粗視化を行うのと同時にMartini 3力場の力場タイプを割り付けできます
古典シミュレーションの反応力場
通常の古典分子動力学法では、分子の結合状態が変わることはありませんが、材料組成の経時的な変化を正確にシミュレーションするためには、化学反応を考慮する必要があります。これを実現する方法の1つに、反応物分子から反応が起こり得る部位を同定し、確率的なルールに従い、仮想的に反応を発生させることを繰り返していく方法が挙げられます。Materials Studio 2022では、Perlスクリプトの機能として実装されたReactionFinder という機能を活用して、反応分子動力学に基づき様々な反応系のシミュレーションが可能です。ReactionFinderの機能により、反応部位の同定や反応による結合状態の変化をPerlスクリプトの一部として実行することができます。この2つの機能は、以下の新しいチュートリアルで確認できます。
- チュートリアルの追加:Executing Virtual Atomistic Reactions with ReactionFinder (ReactionFinderを活用した仮想的な反応の実行)では、ReactionFinderを活用して、仮想的に反応を実行する方法を学ぶことができます。
- チュートリアルの追加:Using ReactionFinder to Simulate the Formation of the Solid Electrolyte Interphase(ReactionFinder を活用した固体電解質界面の形成シミュレーション)では、モンテカルロ法と分子動力学に基づく大規模なハイブリッド・アルゴリズムの一部として、ReactionFinder を活用する方法を説明します。固体電解質界面(SEI)形成における負極電解質界面の複雑かつ長時間にわたる反応の挙動をシミュレーションします。

図7:新しいReactionFinder を使用した、モンテカルロ法と分子動力学のハイブリッド・アルゴリズムに基づく反応分子動力学に基づくシミュレーションの実行例 - リチウムイオン電池の黒鉛負極表面での固体電解質界面の形成
量子力学シミュレーション
量子力学に基づく計算法をベースにしたソルバーでは、以下がアップデートされました。
- パラメータの追加:CASTEP のm-GGAの計算でMBD*の分散力補正を用いた計算で各種のパラメータを指定することが可能になりました。
- 新しい特性:CASTEP のベリー位相法を用いた圧電係数の計算
- 利便性の強化:DMol3のCOSMO溶媒和モデルを使用した計算をPerlスクリプトから行う際に、計算結果を含むハッシュ変数に.cosmo形式のファイルが返されるようになりました
- 精度の向上:DFTB+がバージョン 20.2.1にアップグレードされ、力の計算精度が向上しました
- 設定の追加:DFTB+のスピン分極を考慮する計算で、分子や固体の基底状態のスピン状態の緩和ができるようになりました
- パラメータの追加:GrimmeのD4分散力補正がDFTB+で利用できるようになりました
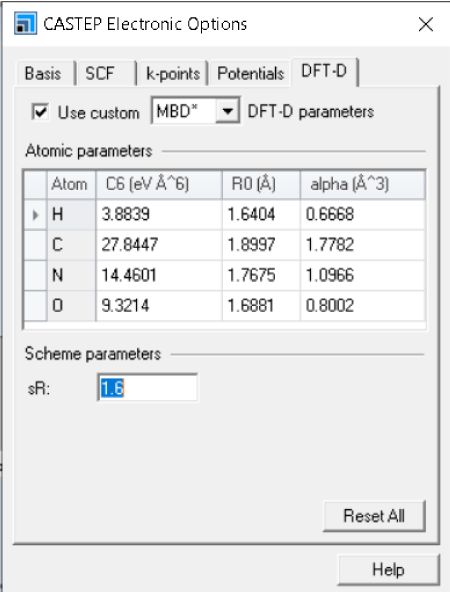
図8:CASTEPのRSCAN汎関数による計算で、MBD*の分散力補正を用いる場合に、GUIから各種のパラメータを指定して計算を実行することができます
サポート対象プラットフォームの変更
サポート対象に加えられたプラットフォーム
- ed Hat Enterprise Linux 8.3 および 8.4に対応しました
- 以下のバージョンのジョブ管理システムに対応しました
- - Altair Portable Batch System(PBS)Pro 2020.x および2021.1.x
- - lurm Workload Manager 20.11.xおよび21.08.x
- 以下のバージョンのブラウザに対応しました
- - Mozilla Firefox ESR 78以降
サポートが終了したプラットフォーム
- CentOS 8.x がサポート対象外となりました
- 以下のバージョンのジョブ管理システムがサポート対象外となりました
- - Slurm Workload Manager 19.05
- - Adaptive Computing TORQUE 6.1.2
- 以下のバージョンのブラウザがサポート対象外となりました
- - Mozilla Firefox ESR 68
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)