- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション  建設業務改善ソリューション
建設業務改善ソリューション  ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER CeeD TOP
FILDER CeeD TOP  FILDER CeeD 電気 TOP
FILDER CeeD 電気 TOP  Rebro D TOP
Rebro D TOP  データ・サイエンス・ソリューション Pipeline Pilot
データ・サイエンス・ソリューション Pipeline Pilot  ライフサイエンス向けソフト Discovery Studio
ライフサイエンス向けソフト Discovery Studio 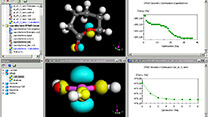 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio  電子実験ノート
電子実験ノート 総合3DCG 制作ソフト Maya
総合3DCG 制作ソフト Maya  総合3DCG 制作ソフト 3ds Max
総合3DCG 制作ソフト 3ds Max  総合3DCG 制作ソフト MODO
総合3DCG 制作ソフト MODO  アニメーション制作ソフト Toon Boom
アニメーション制作ソフト Toon Boom 
分子モデリング・
シミュレーションソフトウェア
Materials Studio
Materials Studio 2023(2022年12月リリース)
- Windows11に対応しました。
- MesociteとForciteで、密度場からの構造因子の計算が可能になりました。
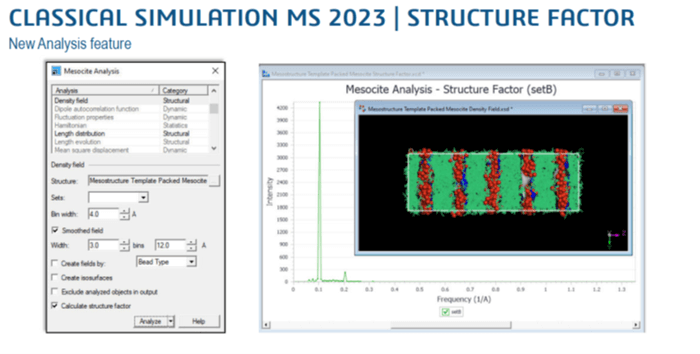
- 振動特性のCASTEP線形応答計算は、スピン偏極(磁気)系に対して実装されました。
- MesociteとForciteで異方性成分による圧力解析がサポートされました。
- CASTEP DFT+Uでは、磁性を持たない系に対してスピン偏極の設定が不要になり、計算が2倍以上速くなりました。
- ForciteのGPU計算がEmbedded Atomに対応しました。
- ForciteダイナミクスGPUアルゴリズムの見直しにより、パフォーマンスが大幅に向上しました。
- CASTEPでは、陰溶媒存在下でのForce計算が可能になり、溶媒の影響を考慮した構造最適化、分子動力学等の実行が可能になりました。
- COMPASSIII水モデルで、拡散係数の予測精度が向上しました。

活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)